Sacando algunas de las conjeturas del descubrimiento de drogas
Un modelo de aprendizaje profundo predice rápidamente las formas tridimensionales de moléculas similares a fármacos, lo que podría acelerar el proceso de descubrimiento de nuevos medicamentos.
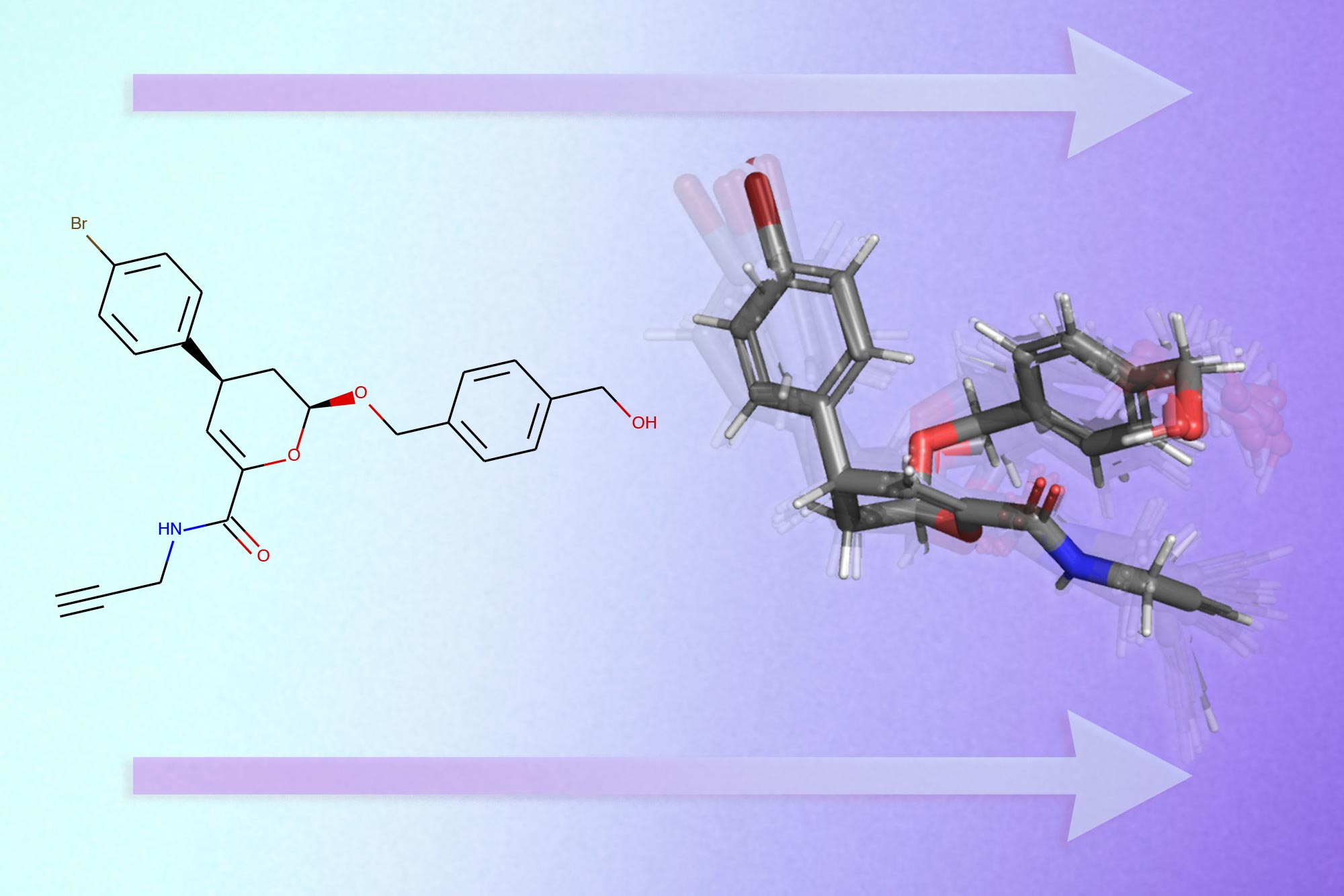
En su búsqueda por descubrir nuevos medicamentos efectivos, los científicos buscan moléculas similares a los medicamentos que puedan unirse a las proteínas que causan enfermedades y cambiar su funcionalidad.
Es crucial que conozcan la forma tridimensional de una molécula para comprender cómo se adherirá a superficies específicas de la proteína.
Pero una sola molécula puede plegarse de miles de formas diferentes, por lo que resolver ese acertijo experimentalmente es un proceso costoso y que requiere mucho tiempo, similar a buscar una aguja en un pajar molecular.
Los investigadores del MIT están utilizando el aprendizaje automático para agilizar esta compleja tarea.
Han creado un modelo de aprendizaje profundo que predice las formas 3D de una molécula basándose únicamente en un gráfico en 2D de su estructura molecular.
Las moléculas se representan típicamente como pequeños gráficos.
Su sistema, GeoMol, procesa moléculas en solo segundos y funciona mejor que otros modelos de aprendizaje automático, incluidos algunos métodos comerciales.
GeoMol podría ayudar a las compañías farmacéuticas a acelerar el proceso de descubrimiento de fármacos al reducir la cantidad de moléculas que necesitan probar en experimentos de laboratorio, dice Octavian-Eugen Ganea, postdoctorado en el Laboratorio de Ciencias de la Computación e Inteligencia Artificial (CSAIL) y coautor principal de el papel.
“Cuando piensas en cómo se mueven estas estructuras en el espacio 3D, en realidad solo hay ciertas partes de la molécula que son realmente flexibles.
Una de las innovaciones clave de este trabajo es en modelar la flexibilidad conformacional como lo haría un ingeniero químico.
Realmente se trata de intentar predecir la distribución potencial de enlaces rotativos en la estructura ”, dice Lagnajit Pattanaik, estudiante de posgrado en el Departamento de Ingeniería Química y coautor principal del artículo.
Otros autores incluyen a Connor W. Coley, profesor asistente de desarrollo profesional de ingeniería química Henri Slezynger; Regina Barzilay, profesora distinguida de IA y salud de la Escuela de Ingeniería en CSAIL; Klavs F. Jensen, Profesor Warren K. Lewis de Ingeniería Química; William H. Green, profesor Hoyt C. Hottel de Ingeniería Química; y el autor principal Tommi S. Jaakkola, profesor Thomas Siebel de Ingeniería Eléctrica en CSAIL y miembro del Instituto de Datos, Sistemas y Sociedad. La investigación se presentará esta semana en la Conferencia sobre Sistemas de Procesamiento de Información Neural.
Mapeo de una molécula
En un gráfico molecular, los átomos individuales de una molécula se representan como nodos y los enlaces químicos que los conectan son bordes.
GeoMol aprovecha una herramienta reciente en aprendizaje profundo llamada red neuronal de paso de mensajes, que está específicamente diseñada para operar en gráficos.
Los investigadores adaptaron una red neuronal de paso de mensajes para predecir elementos específicos de geometría molecular.
Dado un gráfico molecular, GeoMol predice inicialmente las longitudes de los enlaces químicos entre los átomos y los ángulos de esos enlaces individuales. La forma en que se organizan y conectan los átomos determina qué enlaces pueden rotar.
Luego, GeoMol predice la estructura de la vecindad local de cada átomo individualmente y ensambla pares vecinos de enlaces rotativos calculando los ángulos de torsión y luego alineándolos.
Un ángulo de torsión determina el movimiento de tres segmentos que están conectados, en este caso, tres enlaces químicos que conectan cuatro átomos.
“Aquí, los enlaces rotativos pueden tomar una amplia gama de valores posibles. Entonces, el uso de estas redes neuronales de transmisión de mensajes nos permite capturar muchos de los entornos locales y globales que influyen en esa predicción. El enlace rotativo puede tomar varios valores y queremos que nuestra predicción pueda reflejar esa distribución subyacente ”, dice Pattanaik.
Superar los obstáculos existentes
Un desafío importante para predecir la estructura 3D de las moléculas es modelar la quiralidad.
Una molécula quiral no se puede superponer a su imagen especular, como un par de manos (no importa cómo gires las manos, no hay forma de que sus características se alineen exactamente).
Si una molécula es quiral, su imagen especular no interactuará con el medio ambiente de la misma manera.
Esto podría hacer que los medicamentos interactúen incorrectamente con las proteínas, lo que podría resultar en efectos secundarios peligrosos.
Los métodos actuales de aprendizaje automático a menudo implican un proceso de optimización largo y complejo para garantizar que la quiralidad se identifique correctamente, dice Ganea.
Debido a que GeoMol determina la estructura 3D de cada enlace individualmente, define explícitamente la quiralidad durante el proceso de predicción, eliminando la necesidad de optimización después del hecho.
Después de realizar estas predicciones, GeoMol genera un conjunto de estructuras 3D probables para la molécula.
“Lo que podemos hacer ahora es tomar nuestro modelo y conectarlo de un extremo a otro con un modelo que predice este apego a superficies proteicas específicas. Nuestro modelo no es una tubería separada. Es muy fácil de integrar con otros modelos de aprendizaje profundo ”, dice Ganea.
Un modelo “superrápido”
Los investigadores probaron su modelo utilizando un conjunto de datos de moléculas y las posibles formas en 3D que podrían tomar, que fue desarrollado por Rafael Gomez-Bombarelli, el presidente de desarrollo, el ingeniero Jeffrey Cheah, y el estudiante graduado Simon Axelrod.
Evaluaron cuántas de estas estructuras 3D probables pudo capturar su modelo, en comparación con los modelos de aprendizaje automático y otros métodos.
En casi todos los casos, GeoMol superó a los otros modelos en todas las métricas probadas.
“Descubrimos que nuestro modelo es súper rápido, lo cual fue realmente emocionante de ver. Y lo que es más importante, a medida que agrega más enlaces rotativos, se espera que estos algoritmos se desaceleren significativamente. Pero realmente no vimos eso. La velocidad se escala muy bien con el número de enlaces rotativos, lo que es prometedor para el uso de este tipo de modelos en el futuro, especialmente para aplicaciones en las que se intenta predecir rápidamente las estructuras 3D dentro de estas proteínas ”, dice Pattanaik.
En el futuro, los investigadores esperan aplicar GeoMol al área de detección virtual de alto rendimiento, utilizando el modelo para determinar estructuras de moléculas pequeñas que interactuarían con una proteína específica.
También quieren seguir refinando GeoMol con datos de entrenamiento adicionales para que pueda predecir de manera más efectiva la estructura de moléculas largas con muchos enlaces flexibles.
“El análisis conformacional es un componente clave de numerosas tareas en el diseño de fármacos asistido por computadora, y un componente importante en el avance de los enfoques de aprendizaje automático en el descubrimiento de fármacos”, dice Pat Walters, vicepresidente senior de computación en Relay Therapeutics, que no participó en esta investigaciónr. “Estoy emocionado por los continuos avances en el campo y agradezco al MIT por contribuir a un aprendizaje más amplio en esta área”.
Referencia:
“GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles” por Octavian-Eugen Ganea, Lagnajit Pattanaik, Connor W. Coley, Regina Barzilay, Klavs F. Jensen, William H. Green y Tommi S. Jaakkola, 8 de junio de 2021, Física> Física química .
arXiv: 2106.07802